

Risikomanagement und Warnungen:
Wie sich Risiken senken lassen

Dass die Risikoanalyse ein lebendes Dokument ist, sollte spätestens seit der Einführung der EU-Medizinprodukteverordnung 2017/745 (MDR) bekannt sein. Die MDR definiert ein Risiko als: “die Kombination von Wahrscheinlichkeit eines Schadenseintritts und Schwere des Schadens“. Die Risikoanalyse bewertet Risiken, die entweder vor dem Marktstart antizipiert, oder bei der Anwendung im klinischen Alltag bekannt geworden sind. Der Hersteller bewertet auf Basis der Schwere des Schadens und der Wahrscheinlichkeit des Schadenseintritts, ob ein Risiko in Anbetracht des Nutzens akzeptabel erscheint. Nicht akzeptable Risiken müssen zwingend in Schwere oder Wahrscheinlichkeit reduziert werden, ein Design-Change ist jedoch sehr aufwändig. Da erscheint es naheliegend, durch die leichter anpassbare Gebrauchsanweisung über das Risiko aufzuklären und somit die Auftretenswahrscheinlichkeit zu verringern. Wann diese Maßnahme ausreicht, und in welcher Beziehung die Risikoanalyse zur klinischen Bewertung steht, erläutert dieser Artikel.
Warum Risiken reduzieren?
Die MDR (Anhang I, Kapitel 1, Punkt 1) ist hier eindeutig: „Sie [die Produkte] sind sicher und wirksam und gefährden weder den klinischen Zustand und die Sicherheit der Patienten noch […] die Gesundheit der Anwendung oder gegebenenfalls Dritter.“ Und sagt „[…], dass Risiken so weit zu verringern sind, wie es ohne negative Auswirkungen auf das Nutzen-Risiko-Verhältnis möglich ist“. Welche Wege hat ein Hersteller, um Risiken zu reduzieren und welche Anforderungen stellt die MDR?
Hersteller haben ein Eigeninteresse Risiken zu minimieren, da sich sichere Produkte langfristig gegenüber der Konkurrenz durchsetzen. MDR gibt Möglichkeiten an die Hand, Risikoreduzierung zu betreiben.
Risikosenkende Maßnahmen
Mit dem Inkrafttreten der MDR wurde eine wichtige Regel für die risikosenkende Maßnahmen grundlegend verändert. Als Teil der Risikosenkung war es bis dahin nicht möglich, Risiken allein durch einen Warnhinweis, zum Beispiel in einer Gebrauchsanweisung, zu senken.
ISO 14971:2012 und Richtlinie 93/42 EWG
Die ISO 14971:2012 (Risikomanagement für Medizinprodukte) gibt im Anhang ZA, Abschnitt 7, b) an: „, dass die Nutzer über die Restrisiken zu informieren sind. Das weist darauf hin, dass nach Richtlinie 93/42 EWG Anhang l, und im Gegensatz zur Auffassung der Norm die Informationen, die an Nutzer ausgegeben werden, das (Rest-)Risiko nicht weiter vermindern“.
Damit mussten Risiken durch Design oder Schutzmaßnahmen minimiert werden. Rest-Risiken, die nicht weiter gesenkt werden konnten, mussten und müssen nach wie vor durch den Nutzen aufgewogen werden und wurden in den Warnhinweisen angesprochen.
Was sich mit der MDR ändert
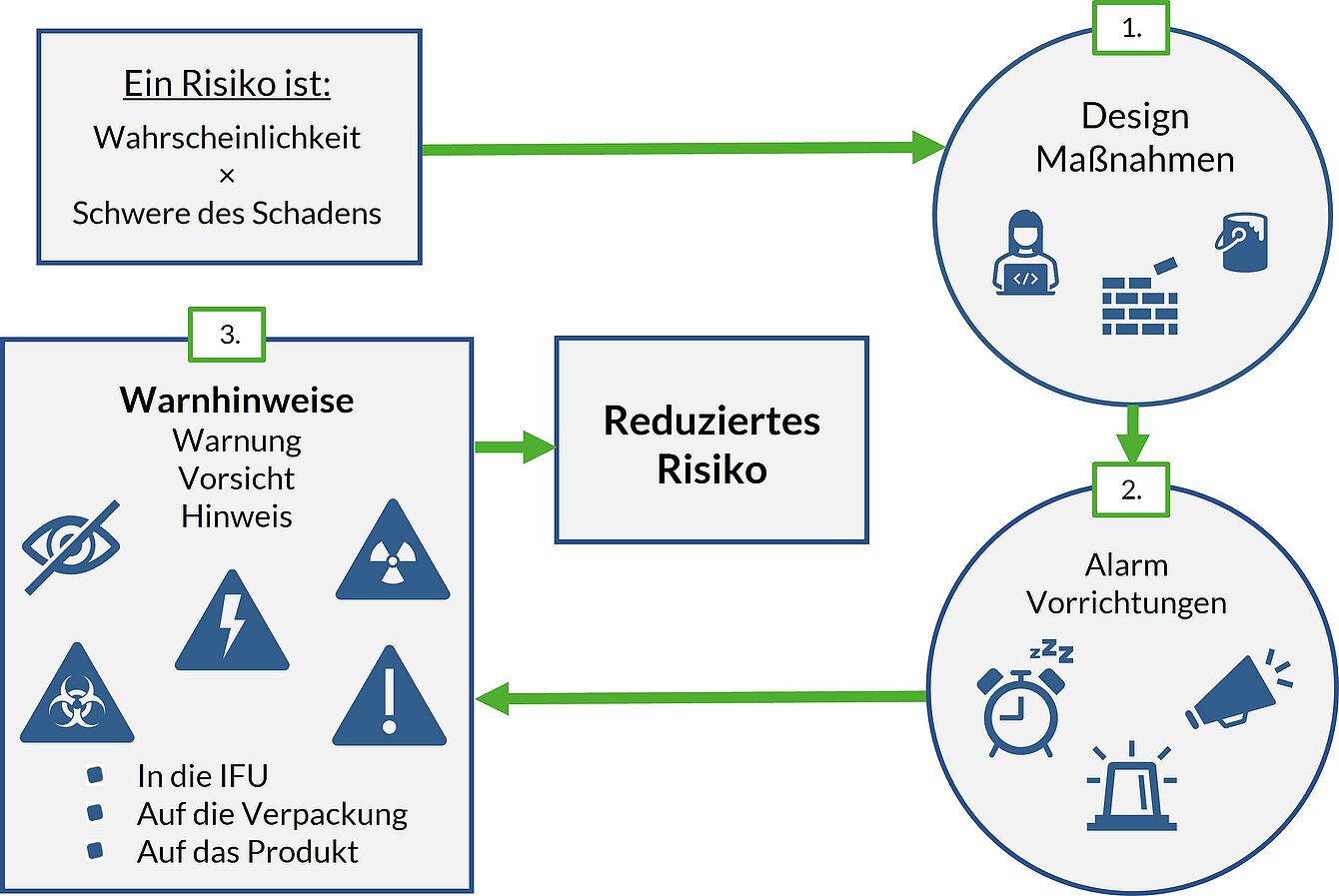
Die gute Nachricht: Unter der MDR können Risiken über Warnhinweise und Informationen in der Gebrauchsanweisung gemindert werden. Aber dazu muss noch immer die Rangfolge der risikosenkenden Maßnahmen beachtet und dieser Schritt im Zweifel begründet werden. Die MDR, Anhang I, Punkt 4 dazu:
„Bei der Wahl der am besten geeigneten Lösungen müssen die Hersteller in nachstehender Rangfolge
- die Risiken durch sichere Auslegung und Herstellung beseitigen oder so weit wie möglich minimieren,
- gegebenenfalls angemessene Schutzmaßnahmen, soweit erforderlich einschließlich Alarmvorrichtungen, im Hinblick auf nicht auszuschließende Risiken ergreifen und,
- Sicherheitsinformationen (Warnungen, Vorsichtshinweise, Kontraindikationen) sowie gegebenenfalls Schulungen für Anwender bereitstellen.“
Die Rangfolge der Maßnahmen
Das bedeutet, Risiken müssen vorzugsweise durch Designmaßnahmen gesenkt werden, zum Beispiel indem ein Herzschrittmacher eine besonders langlebige Batterie erhält, um einen Funktionsverlust zu vermeiden. Ist das Risiko danach noch immer inakzeptabel, müssen Schutzmaßnahmen ergriffen werden, zum Beispiel indem der Herzschrittmacher einen Alarm gibt, bevor die Batterie aufgebraucht ist. Erst wenn das Risiko nicht weiter gesenkt werden kann, darf ein Hersteller einen Sicherheitshinweis in der Gebrauchsanweisung zur Risikosenkung nutzen, zum Beispiel mit dem Hinweis, dass die Batterie eine begrenzte Lebenszeit besitzt.
Ob ein Hersteller alle Design- und Schutzmaßnahmen ausgeschöpft hat, und ob diese ausreichen, ist individuell zu betrachten. Hersteller sollten sich auf Nachfragen der Benannten Stelle einstellen und ihre Vorgehensweise begründen können.
Viel hilft nicht viel
Wichtig ist es, diese Art der Risikosenkung nicht durch viel zu viele Hinweise und Warnungen zu überstrapazieren.
Hier äußert sich die ISO/TR 24971: 2020 in Kapitel 8.2, d) ganz klar: “An analysis might be needed to determine if there is an over-reliance on warnings”.
Warnhinweise sollen also verantwortungsvoll eingesetzt werden. Ob die Hinweise auch die gewünschte Wirkung erzielen, kann zum Beispiel über Usability-Tests überprüft werden.
Warnhinweise leicht gemacht
Wenn Warnhinweise eingesetzt werden, sollte ihre Formulierung und Form mit Bedacht gewählt werden. Bisher gibt es noch keine harmonisierte Norm zum Thema Warnhinweise. Auch die MDR thematisiert diese wenig. Sie nennt im Anhang I, Abschnitt 23 folgende Punkte: „Medium, Format, Inhalt, Lesbarkeit und Anbringungsstelle der Kennzeichnung und der Gebrauchsanweisung eignen sich für das jeweilige Produkt. […] Insbesondere ist die Gebrauchsanweisung so zu verfassen, dass sie von dem vorgesehenen Anwender ohne Schwierigkeiten verstanden wird.“
Da keine MDR-Vorgaben existieren wird von manchen Benannten Stellen auf Normen zurückgegriffen. Unsere Erfahrung zeigt hier, dass zum Beispiel die IEC 82079-1:2019, Teil 1, welche die Erstellung von Nutzungsinformationen regelt, von einigen Benannten Stellen als relevant betrachtet wird, obwohl sie sich nicht speziell auf Medizinprodukte bezieht.
So sind die Informationen zu „Schäden an Personen“ mit den Signalwörtern Gefahr, Warnung und Vorsicht zu kennzeichen. Für Sachschäden sollen die Begriffe Hinweis oder Achtung verwendet werden. Es gibt keine; Vorschrift zu Farben, jedoch sollen diese in sich konsistent verwendet werden.
Gefahr, Warnung, Vorsicht, Hinweis
Adaptiert aus IEC 82079-1: 2019, Teil 1:
Gefahr: Gefährdungssituation die sicher zum Tod oder zu einer schweren Verletzung führt, wenn nicht vermieden
Warnung: Gefährdungssituation, die vielleicht zum Tod oder zu einer schweren Verletzung führt, wenn nicht vermieden.
Vorsicht: Gefährdungssituation, die vielleicht zu geringfügiger oder mäßiger Verletzung führt, wenn nicht vermieden
Hinweis oder Warnung: Gefährdungssituation die zu Sachschaden am Produkt (oder anderem Eigentum) führen kann, wenn nicht vermieden
Was ist für die klinische Bewertung zu beachten?
Die Risikoanalyse und die Warnhinweise in der Gebrauchsanweisung werden auch in die klinische Bewertung einbezogen. Dabei werden die genannten Risiken sorgfältig mit den klinischen Daten aus der Literatur verglichen:
- Decken sich die identifizierten Risiken in Bezug auf Schwere und Wahrscheinlichkeiten eines Schadens mit den Erkenntnissen der Literatur und der Überwachung nach dem Inverkehrbringen?
- Sind eventuell Kontraindikationen in der Literatur beschrieben, die nicht in der Gebrauchsanweisung abgebildet sind?
- Ist die Formulierung und Wahl der Kontraindikationen schlüssig?
- Ist die Auftretenswahrscheinlichkeit eines Risikos sinnvoll bemessen?
Mit Blick auf die IFU wird geprüft, ob Warnhinweise, die in der Risikoanalyse zur Risikosenkung festgelegt werden, in der IFU umgesetzt sind.
Zudem wird geprüft, ob Risiken sinnvoll mit Blick auf die Art des Medizinprodukts identifiziert wurden.
Fazit
- Hersteller müssen bei der Risikominimierung zunächst Design- und Schutzmaßnahmen erwägen, bevor sie einen Warnhinweis als risikosenkende Maßnahme einsetzen können.
- Werden Warnhinweise verwendet, um das Risiko zu senken, könnte ein Usability-Test die Wirksamkeit der Maßnahme nachweisen.
- Warnhinweise können zum Beispiel nach der IEC 82079-1: 2019 erstellt werden. Zwar ist diese Norm bisher nicht harmonisiert und gilt für alle Arten von Gebrauchsanweisungen (nicht speziell für Medizinprodukte), gibt den Herstellern jedoch eine gute Hilfestellung. Da die Vorgängernorm harmonisiert war, ist es durchaus möglich, dass auch diese Version harmonisiert wird.
- In der Durchführung der klinischen Bewertung werden Warnhinweise als risikosenkende Maßnahme in ihrer Umsetzung überprüft.
Kontakt
Dominik Heiss, B.Sc.
dominik.heiss@novineon.com
+49 7071 / 98979 - 150
Download

Dominik Heiss, B.Sc.
Consultant
dominik.heiss@novineon.com
+49 7071 / 98979 - 150