

In-Vitro Diagnostic Medical Devices Regulation (EU) 2017/746
Übergangsfristen und Anforderungen

Seit 26. Mai 2022 gilt offiziell die IVDR, die IVDD ist damit abgelöst. Um den Übergang aber möglichst reibungslos zu gestalten, gibt es unterschiedlich lange Übergangsfristen für die Produkte der verschiedenen Risikoklassen. Darüber hinaus gelten manche IVDR-Anforderungen für Produkte, die vor oder während des Übergangszeitraumes noch unter IVDD zertifiziert wurden, schon jetzt in der Übergangsphase. Wie diese Anforderungen aussehen und wann eine Mitteilung an die benannte Stelle nötig ist, diskutiert Timo Brüggemann in diesem Artikel.
Die EU-Verordnung 2017/746 zu In-vitro-Diagnostika (IVDR) hat am 26. Mai 2022 die Richtlinie 98/79/EG IVDD abgelöst. Die Anforderungen der IVDR gelten für alle Produkte, die ab dem 26. Mai 2022 in Verkehr gebracht oder in Betrieb genommen werden.
Gemäß diesen Anforderungen muss für das Konformitätsbewertungsverfahren der Mehrzahl der Produkte nun eine benannte Stelle einbezogen werden. Die Anzahl der für eine Konformitätsbewertung unter der IVDR benannten Stellen ist bislang jedoch gering. Um dennoch für einen reibungslosen Übergang zu sorgen und die Verfügbarkeit von Produkten auf dem europäischen Markt sicherzustellen, gewährt die EU-Kommission den Herstellern verlängerte Übergangsfristen (Verordnung (EU) 2022/112). Darüber hinaus entfällt nun ebenfalls die bis Anfang des Jahres geltende Abverkaufsfrist (Verordnung (EU) 2023/607), wobei die jeweilige Haltbarkeit der Produkte hierbei zu beachten ist.
Verlängerung der Übergangszeiträume
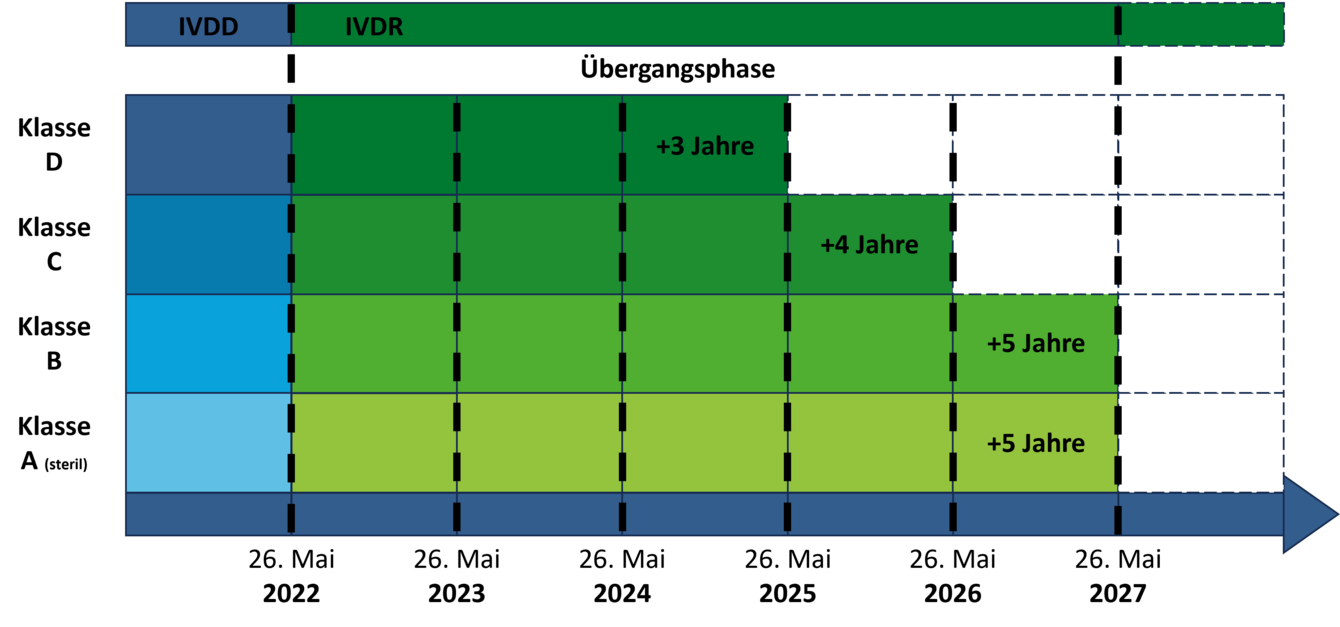
Die Verlängerung der Übergangszeiträume beschränkt sich auf Bestandsprodukte, für die bereits vor dem 26. Mai 2022 eine Konformitätserklärung ausgestellt wurde. Die Verlängerung ist abhängig von der Risikoklasse des Produkts und davon, ob für die Konformitätsbewertung unter IVDD bereits eine benannte Stelle involviert war.
Für Produkte, für welche bereits unter der IVDD die Beteiligung einer benannten Stelle gefordert war (Liste A+B und Produkte zur Eigenanwendung), wird die Übergangszeit um ein Jahr, bis zum 26. Mai 2025, verlängert.
Für Produkte, welche für die Konformitätsbewertung nach IVDR erstmalig eine benannte Stelle benötigen, sind die Übergangszeiträume nun folgendermaßen definiert:
- Klasse D-Produkte: 26. Mai 2025
- Klasse C-Produkte: 26. Mai 2026
- Sterile Klasse A-Produkte und Klasse B-Produkte: 26. Mai 2027
- Für sogenannte Inhouse-Tests verlängert sich die Frist bis 26. Mai 2028, sofern kein vergleichbares Produkt auf dem Markt zugelassen ist.
Unsterile Klasse A-Produkte sowie neue Produkte, für welche erst nach dem 26. Mai 2022 eine Konformitätserklärung ausgestellt wird, sind von den verlängerten Übergangsfristen ausgeschlossen.
Überwachungspflichten und Vigilanz auf IVDR-Niveau
Ungeachtet dieser Fristenverlängerung müssen Hersteller jedoch ihren Pflichten hinsichtlich der Überwachung der Produkte im Markt und Vigilanz nachkommen, auch wenn die Produkte noch unter IVDD in Verkehr gebracht werden.
Das im Mai 2022 veröffentliche MDCG-Dokument 2022-8 gibt Hilfestellung, wie die Anforderungen der IVDR für Bestandsprodukte umzusetzen sind. Im MDCG-Dokument erfolgt eine Unterteilung von Bestandsprodukten in ‚Old‘ und ‚Legacy‘ Devices, diese Begrifflichkeiten werden in der IVDR aber nicht genannt und finden lediglich in MDCG-Dokumenten Gebrauch.
Je nachdem ob es sich bei einem Bestandsprodukt um ein ‚Old‘ oder ein ‚Legacy‘ Device handelt, gelten bis zu den genannten Stichtagen verschiedene Anforderungen.
‚Legacy‘ Devices
‚Legacy‘ Devices sind Produkte, zwischen 26.Mai 2022 und dem oben genannten jeweiligen Ende der Übergangszeit in Verkehr gebracht werden, sofern diese keine signifikanten Änderungen hinsichtlich Designs und Zweckbestimmung erfahren. Dies können Produkte sein, für die schon vor dem 26. Mai 2022 ein Zertifikat von einer benannten Stelle gemäß IVDD ausgestellt wurde, oder Produkte, für die vor dem 26. Mai 2022 eine Konformitätserklärung gemäß IVDD ohne Beteiligung einer benannten Stelle erstellt wurde und für welche unter IVDR nun das Mitwirken einer benannten Stelle gefordert ist.
Für ‚Legacy‘ Devices gelten außerdem Anforderungen der IVDR an die Marktüberwachung (PMS) und Vigilanz (Kapitel VII, IVDR). Es wird zwingend ein auf einem PMS-Plan basierendes PMS-System gefordert, sowie ein Post-Market Performance Follow-Up (PMPF), das Teil dieses PMS-Systems ist. Unabhängig von der Risikoklasse benötigt jedes Legacy Device einen PMS-Bericht (Art.80, IVDR), sollte es sich um ein Produkt der Klasse C oder D handeln, kann stattdessen freiwillig ein periodic safety update report (PSUR) vorbereitet werden (Art.81, IVDR).
Zusätzlich haben die Wirtschaftsakteure von ‚Legacy‘ Devices Verpflichtungen, die sie erfüllen müssen, dies betrifft Hersteller (Art.11, IVDR), autorisierte Repräsentanten (Art.11, IVDR), Importeure (Art.13, IVDR) und Distributoren (Art.14, IVDR). Prinzipiell muss auch die Registrierung von Produkten (Art.26, IVDR) und die Registrierung von Wirtschaftsakteuren (Art.28, IVDR) konform mit der IVDR sein. Da EUDAMED altuell noch nicht voll funktionsfähig ist, gelten besondere Übergangsbestimmungen die in Artikel 112 und 113(3) der IVDR beschrieben sind.
‚Old‘ Devices
Zu ‚Old‘ Devices werden Produkte gezählt, welche vor dem 26. Mai 2022 in Verkehr gebracht wurden und für welche ein gültiges Zertifikat gemäß IVDD vor dem 26. Mai 2022 oder den geltenden nationalen Bestimmungen vor dem Inkrafttreten der IVDD ausgestellt wurde. Für diese Produkte greift die IVDR per se nicht, jedoch müssen Hersteller hier ebenfalls ihrer Pflicht hinsichtlich der Marktüberwachung der Produkte nachkommen. Das bedeutet, dass nach dem 26. Mai 2022 auch die Artikel zur Marktüberwachung (Art.88 bis 95, IVDR) durch die zuständigen Behörden gelten. Um weiter zu gewährleisten, dass alle Berichte und Analysen von schwerwiegenden Vorfällen und Sicherheitskorrekturmaßnahmen im Feld (FSCAs) im Zuge der PMS verarbeitet werden, gelten außerdem die Artikel zu schwerwiegenden Vorkommnissen und daraus resultierender Maßnahmen (Art.82 und 84, IVDR).
Geltende IVDR-Anforderungen während des Übergangszeitraumes für ‚Legacy‘ und ‚Old‘ Devices.
| 'Legacy device' | 'Old device' |
|---|---|
| Verpflichtungen von Herstellenden, Repräsentativen, Importeuren und Verteilern (Art.10, 11, 13 und 14, IVDR) | |
| Registrierung von Produkten und Wirtschaftsakteuren (Art. 26 und 28, IVDR) in Übereinstimmung mit Art.112 und 113(3) | |
| PMS System mit PMS-Plan (Art. 78, 79, IVDR) | |
| PMS Report (Art. 80) ODER ein PSUR (Art. 81, IVDR) | |
| Vigilanz (Art. 82 bis 87, IVDR) | Schwerwiegende Vorkommnisse und daraus resultierende Maßnahmen (Art. 82 und 84, IVDR) |
| Marktüberwachung (Art. 88 bis 95, IVDR) | Marktüberwachung (Art. 88 bis 95, IVDR) |
Was, wenn es Änderungen am Produkt im Übergangszeitraum gibt?
All diese Fristverlängerungen und die beschränkten IVDR-Anforderungen für ‚Legacy‘ Devices gelten aber nur für den Fall, dass es keine signifikanten Änderungen an den Produkten in Bezug auf Zweckbestimmung und Design während der Übergangsfrist gab.
Wenn es Änderungen gibt, muss erfasst werden, ob diese signifikant waren. Handelt es sich um nicht-signifikante Änderungen, kann das Produkt während der Übergangsfrist weiter unter IVDD zertifiziert bleiben, bis die jeweilige Übergangsfrist ausläuft.
Handelt es sich um signifikante Änderungen, kann das Produkt nicht weiter unter der IVDD auf dem Markt bleiben und muss unter IVDR-Auflagen rezertifiziert werden. Die jeweils zuständigen benannten Stellen verifizieren und dokumentieren die Veränderungen schriftlich und entscheiden im Laufe ihrer Überwachungsarbeit über den Verbleib der Validität.
Das ebenfalls im Mai veröffentlichte MDCG-Dokument 2022-6 gibt Hilfestellung, welche Änderungen als signifikant gelten und welche nicht. Generell werden nur bestimmte Änderungen an der Zweckbestimmung und dem Design als signifikant betrachtet.
Änderungen der Zweckbestimmung
Als signifikante Änderung der Zweckbestimmung wird zum Beispiel eine Erweiterung der Zweckbestimmung oder andere wichtige Änderungen (wie etwa vorgesehene Anwender oder Änderungen der Arbeitsweise) betrachtet. Lediglich die Limitierung der Zweckbestimmung gilt als nicht-signifikante Änderung. Die Zweckbestimmung darf also eingeschränkt aber nicht erweitert oder abgeändert werden.
Änderungen am Design
Signifikante Änderungen am Design sind Änderungen am Funktionsprinzip oder Änderungen, die zu einer Be-
einträchtigung des Risiko-Nutzen-Verhältnisses führen, selbst wenn diese nicht das Funktionsprinzip verändern.
Zum Überbegriff des Designs zählen ebenso Software, Inhaltsstoffe und Materialien, sowie die Sterilisation.
Liegen keine signifikanten Änderungen am Design vor, muss dennoch überprüft werden, ob es Änderungen an Software, Inhaltsstoffen / Materialien oder Sterilisation gibt, die eine signifikante Änderung des Produktes bedeuten. Ergeben all diese Prüfungen keine signifikanten Änderungen, kann das Produkt weiterhin als ‚Old‘ oder als ‚Legacy‘ Device betrachtet werden. Sollte es eine signifikante Änderung geben, dann muss das Produkt unter der IVDR neu zertifiziert werden.
Fazit
Zusammenfassend lässt sich sagen, dass sich für Produkte aller Klassen die Übergangsfristen verlängert haben, je nach Risikoklasse (ausgenommen unsterile Klasse A-Produkte) unterschiedlich lange. Marktüberwachung und Vigilanz müssen dennoch auch für unter der IVDD zertifizierte ‚Old‘ und ‚Legacy‘ Devices schon auf IVDR-Niveau stattfinden. Sollte es im Übergangszeitraum signifikante Änderungen in Bezug auf die Zweckbestimmung oder das Design der ‚Old‘ oder ‚Legacy‘ Devices geben, muss das Produkt unter der IVDR neu zertifiziert werden.
Kontakt
Timo Brüggemann
timo.brueggemann@novineon.com
+49 7071 / 98979 - 201

Timo Brüggemann
Consultant
timo.brueggemann@novineon.com
+49 7071 / 98979 - 140