


Many considerations have led to the development of the Medical Devices Regulation (EU) 2017/745 (MDR). Transparency and the protection of public and patient safety are prominently mentioned. In order to keep pace with scientific and technical developments and overcome divergent interpretations, the EU has established Expert Panels. These can be consulted by Notified Bodies in the process of conformity assessment of certain medical devices. What is an Expert Panel? When does a device has to go through the consultation process? What is the role of the clinical evaluation in this process? These questions are answered in this article.
Which products can be affected?
The Clinical Evaluation Consultation Procedure (CECP) is a procedure described in the MDR (Article 54 and Annex IX, Section 5.1). It is intended as part of the conformity assessment for certain devices, namely:
- Class III implantable devices and
- Class IIb active devices intended to administer and/or remove a medicinal product from the body as described in Annex VIII, Section 6.4 (Rule 12), e.g. dialysis systems.
Hereafter, we refer to these products as "CECP products." The procedure can be initiated for initial MDR certification of existing products or new products, as well as for MDR recertifications, provided that relevant changes have been made to the product.
When is a CECP not required?
There are exceptions for CECP products in which the consultation procedure is not required. These are, according to Art. 54(2) MDR:
- When a certificate issued in accordance with this Regulation [MDR] is renewed
► MDR recertifications are meant here
- where the device has been designed by modifying a device already placed on the market by the same manufacturer for the same intended purpose, provided that the manufacturer has demonstrated to the satisfaction of the Notified Body that the modifications do not affect the risk-benefit ratio of the device,
► This refers to legacy devices that are transferred to the MDR largely unchanged.
- The principles of clinical evaluation of the relevant device type or category have been laid down in a specification according to Article 9 and the Notified Body confirms that the clinical evaluation of this device by the manufacturer is in accordance with the relevant specification for the clinical evaluation of this type of device.
► Relevant specifications (eng. "Common Specifications") exist for the CECP product concerned.
For the interpretation of Article 54 MDR, there is an additional MDCG 2019-3 document. Here it is stated that legacy devices fall under this exception according to Article 54(2b) MDR. Likewise, the type and scope of the changes are addressed there. According to this, only those changes are relevant which are necessary to comply with the requirements under the MDR. This includes, for example, changes in the PMS system in consideration of the requirements for the Summary of Safety and Clinial Performance (SSCP), or changes to the labeling in order to fulfill the corresponding basic safety and performance requirements.
What factors can trigger a CECP for legacy devices?
Although legacy devices are mostly exempt under Art. 54(2b), the following aspects can trigger a CECP for these devices as well:
- Changes or expansions in intended purpose and/or indications.
- Expansion of the patient population
- Addition of sizes or variants that were not covered by prior certification under MDD
- Profound changes in clinical procedures or surgical technique
- Changes to the device and/or its accessories that require evaluation of additional clinical data.
What does the CECP involve?
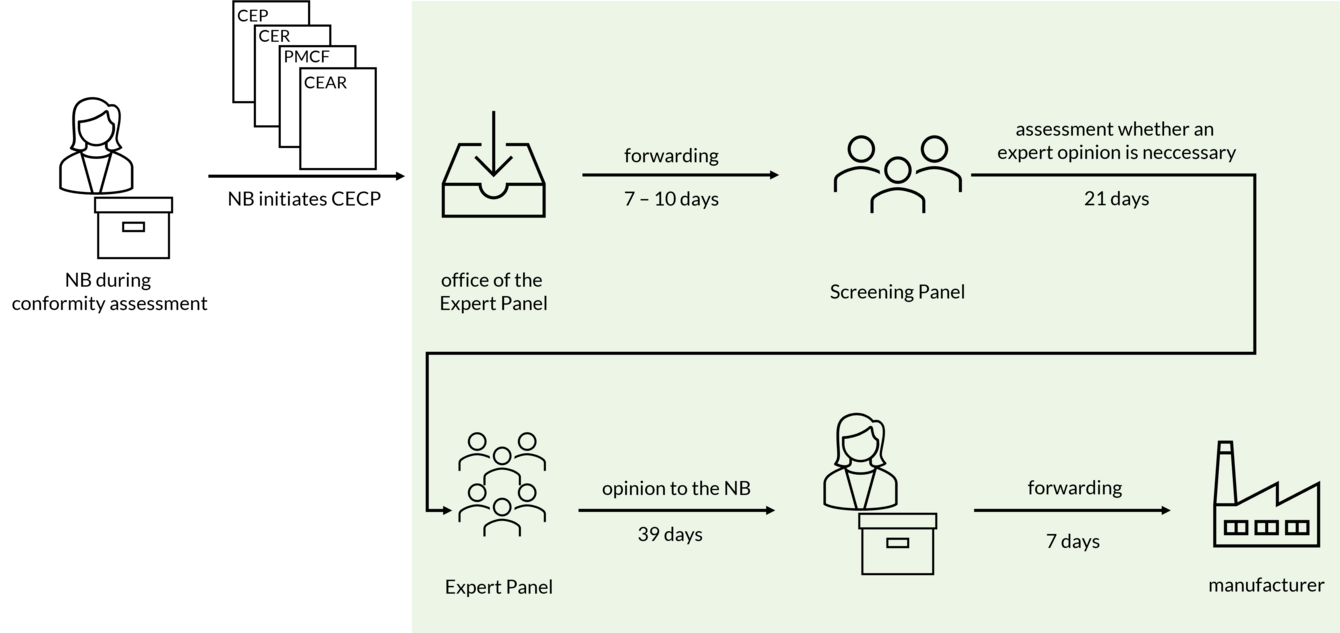
The consultation procedure is initiated quite late in the conformity assessment process. The Notified Body informs the competent authorities whether the CECP is to be applied (Art. 54(3) MDR). The Notified Body must document its decision and make it available to the competent authorities via EUDAMED.
If the Notified Body decides to initiate the CECP, the following documents are forwarded to the office of the Expert Panels:
- Clinical Evaluation Plan (CEP),
- Clinical Evaluation Report (CER),
- PMCF Plan,
- PMCF Report and
- Clinical Evaluation Assessment Report (CEAR).
After 7 to 10 days, this submission is forwarded to the Screening Panel. The Screening Panel decides within 21 days whether an opinion by a thematic expert pannel is necessary for the case at hand. Criteria considered by the Screening Panel are, according to section 5.1c of Annex IX MDR:
- Novelty of the device in question or the associated clinical procedure and its potential significant clinical or public health impact;
- significant adverse change in the benefit-risk profile of a device group due to scientifically substantiated health concerns related to their components or source material or related to the health effects in case of failure of the device
- Significantly increased incidence of serious incidents according to Article 87 in the device group.
If the Screening Panel comes to the decision that a scientific opinion should be provided, the information is forwarded to the appropriate expert panel. The opinion is then submitted to the Notified Body within 39 days and must be forwarded by the Notified Body to the manufacturer within 7 days.
Thus, according to the MDR, it takes 74 - 77 days for the manufacturer to receive the information from the CECP process.
Expert panels
There are 11 expert panels for different disciplines, each of which may have subgroups for specific subfields. The members of the panels are each experts in their field appointed by the European Commission on the basis of their scientific, clinical and technical expertise following a call for expressions of interest. The selection and appointment is made by the European Commission in consultation with the MDCG.
The composition of the expert panels and the published opinions are available on the website of the European Commission.
What effect can the CECP have?
The recommendations of the expert panels are not binding for the Notified Bodies. The expert opinion is published on EUDAMED together with the decision of the Notified Body.
Based on the opinion, the Notified Body can restrict the intended purpose, limit the duration or scope of the certificate, or request adjustments to the technical documentation.
To-dos for the clinical evaluation of legacy devices
Although there is an exemption for legacy devices under Article 54(2b) MDR, there are still some issues that may trigger a CECP as part of the conformity assessment process. This is especially true for changes to the device that affect safety. Since the screening panel decides whether to seek an expert opinion, it is recommended to provide relevant information to the screening panel already in the clinical evaluation.
The clinical evaluation should clearly identify changes to the product or to the definition of the intenden purpose with respect to the above triggers, justify the introduction of these changes, discuss the changes in a risk-based manner, and describe risk-minimizing measures if necessary.
► These basic features of change management reinforce the links of QMS and clinical evaluation and help the Notified Body (and the expert panel) to make appropriate decisions in the CECP.
In addition, the clinical evaluation should include a discussion of the innovations, significant changes in the benefit-risk ratio, and, if applicable, the occurrence of serious incidents according to MDR Annex IX, Section 5.1 c (see above).
This discussion can facilitate the work of the Notified Body and the Screening Panel and should also be considered for initial certifications.
Conclusion
The CECP is a procedure described in the MDR within the conformity assessment for certain class III and IIb devices. An expert panel may be involved by the Notified Body in the conformity assessment procedure. The expert panel may submit a scientific opinion, which will be considered by the Notified Body in the conclusion of the conformity assessment procedure. Legacy devices may also be affected by the CECP. Criteria for initiating the CECP as well as criteria for submitting a scientific opinion are primarily innovations, significant changes in the benefit-risk ratio, and the increase in the rates of serious events. These aspects should be addressed in the clinical evaluation of potentially affected products (legacy devices as well as new products) to facilitate the necessary decisions in the CECP process.

Amko Groeneveld, B.Sc.
Senior Consultant
amko.groeneveld@novineon.com
+49 7071 / 98979 - 147