

FDA-Gruppierung von Medizinprodukten verstehen
Finden Sie sich auf den Webseiten der FDA zurecht

Während in Europa die Produktklassifizierung dem Regelkatalog aus MDR Anhang VIII folgt, so sucht man in den Unterlagen der FDA vergebens nach einem Analogon. novineon CRO unterstützt Medizinproduktehersteller auf Ihrem Weg auf den amerikanischen Markt. In diesem Artikel beschreibt Amko Groeneveld, was bei der Produkteingruppierung zu beachten ist.
Produktklassifizierung
Die US-amerikanische Food and Drug Administration ist die zentrale Bundesbehörde zur Zulassung von Medizinprodukten in den USA. Die jeweiligen Zulassungswege (im Wesentlichen 510(k), DeNovo, PMA) sind durch FDA-eigene Schulungsmaterialien hinreichend gut beschrieben und sollen nur am Rande dieser Fachinformation vorkommen.
Stattdessen konzentrieren wir uns hier auf die Gruppierungsebenen, die die FDA für Medizinprodukte anwendet, und welche Informationen Sie daraus für ihre eigenen Medizinprodukte herauslesen können.
Zunächst der Vergleich mit den europäischen Regularien: Formal beschreibt die MDR 4 Risikoklassen (I, IIa, IIb, III) und gibt im Anhang VIII einen Katalog an Klassifizierungsregeln vor. Ergänzt werden diese Informationen durch MDCG 2021-24, welches zu den vorgenannten Klassifizierungsregeln Beispiele und Anwendungshinweise bereithält.
In den USA begegnet uns ebenfalls eine risikobasierte Klassifizierung (I, II, III). Einen allgemeingültigen Regelkatalog sucht man jedoch vergeblich.
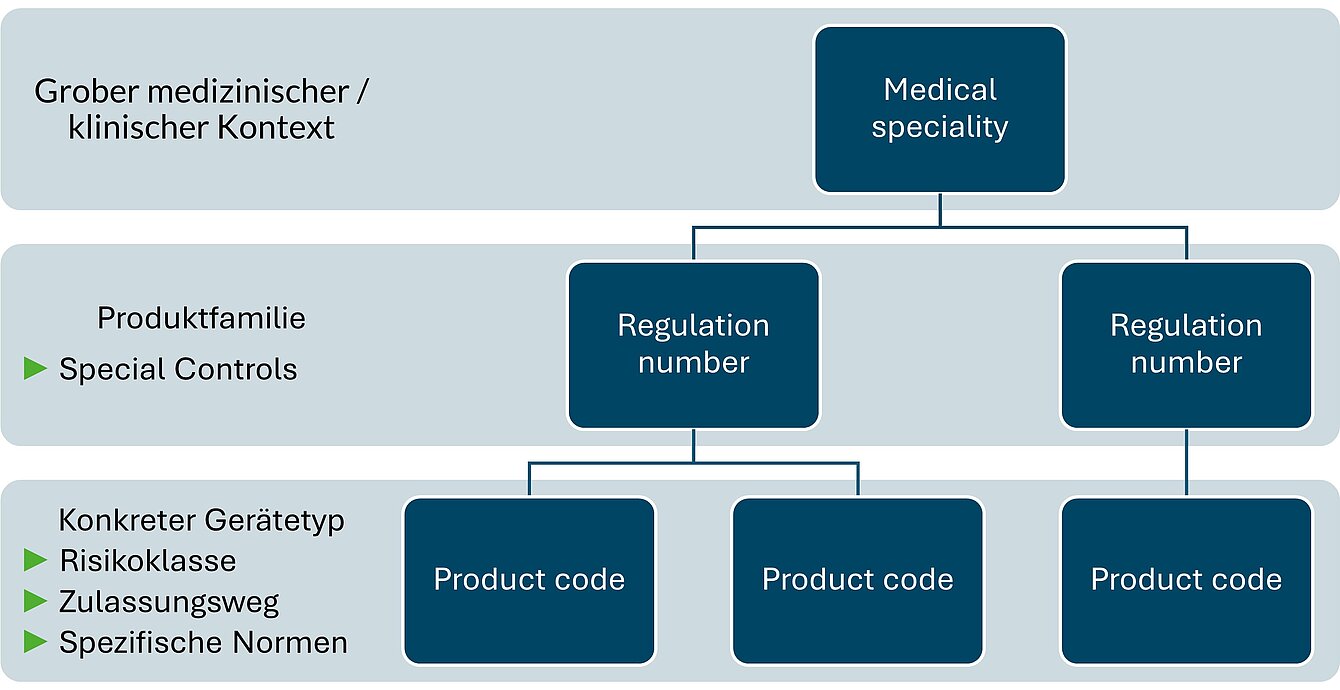
Die FDA hat hier schlicht einen anderen Ansatz zur Gruppierung von Medizinprodukten gewählt. Die Einteilung findet auf drei Ebenen statt:
- Medical Specialty (gröbste Ebene)
- Regulation Number (mittlere Ebene),
- Product Code (spezifischste Ebene)
Erst auf Ebene des Product Codes wird ersichtlich, welche Risikoklasse und welcher Zulassungsweg (bspw. 510(k)) für ein Produkt zutreffend sind. Zum Beispiel können Klasse I und II Produkte bei der FDA eine 510(k) benötigen oder 510(k) exempt sein.
Daher steht eine systematische Recherche des Product Codes am Anfang jedes Einreichungsvorhabens in den USA. Im Folgenden werden diese Ebenen weiter betrachtet und illustriert. Abschließend werden wir Ihnen beispielhaft Ansätze zur Navigation dieser Ebenen aufzeigen, damit Sie für ihre eigenen Produkte mögliche Product Codes – und damit die Risikoklasse und Zulassungswege – identifizieren können.
Medical Specialty
Medical Specialties oder Device Classification Panels bilden einen bestimmten klinisch-medizinischen Bereich ab. Derzeit sind 19 Medical Specialties definiert. Jede dieser Medical Specialties ist in einem eigenen Teil des Titel 21 Code of Federal Regulations (CFR), also des US-Bundesrechts für Medizinprodukten abgebildet, bspw.: „Cardiovascular“ (21 CFR 870), „Gastroenterology and Urology“ (21 CFR 876), oder auch „Ear, Nose, and Throat“ (21 CFR 874). Auf dieser Ebene sind zunächst keine weiteren konkreten regulatorischen Informationen zum eigenen Produkt zu erkennen. Es findet eher eine Orientierung statt – in welchem medizinischen-klinischen Fachbereich bewegt sich mein Produkt?
Je nach Produkt kann der Fachbereich auch uneindeutig sein – Es ist sinnvoll, die Recherche hier aus mehreren Blickwinkeln anzugehen.
Die Medical Specialties finden Sie auf den Seiten der FDA. Beim Klick auf den verlinkten Teil des 21 CFR gelangt man automatisch zur nächsten Ebene welcher wir uns nun widmen wollen – den Regulation Numbers.
Regulation Number
Zu jeder Medical Specialty finden sich neben einleitenden / administrativen Bestimmungen eine ganze Reihe an Regulation Numbers. Diese greifen den Teil des Code 21 CFR der jeweiligen Medical Specialty auf, bspw. 21 CFR 870 für „Cardiovascular“, und fügen eine vierstellige Zahl hinzu. So ist zum Beispiel 21 CFR 870.3925 die Regulation Number für „Replacement Heart Valve“. Eine Regulation Number beschreibt eine Art Produktfamilie innerhalb des medizinisch-klinischen Kontexts (der Medical Specialty).
Auf Ebene der Regulation Number sind eine Beschreibung der Produktfamilie, eine Klassifizierung und ggf. sogenannte Special Controls zu finden. Special Controls sind spezifische Anforderungen an Produkte innerhalb der Regulation Number (Produktfamilie), die diese spezifisch zu erfüllen haben. Diese können in eigenen FDA Guidance Dokumenten beschrieben sein (bspw. für Zahnimplantate) oder direkt in der Regulation Number angegeben werden. Die Special Controls beschreiben unter Anderem bestimmte Punkte zur Untersuchung in Benchtests, Tierversuchen, oder klinischen Studien, oder machen produktspezifische Vorgaben, wie zum Beispiel für das Labeling.
Product Code
Unter jeder Regulation Number sind ein bis mehrere Product Codes angelegt. Stellt man sich die Regulation Number als Produktfamilie vor, dann sind Product Codes die Mitglieder dieser Familie.
Die Größe dieser „Familie“ kann dabei stark variieren. Beispielsweise hat die Regulation Number 876.1500 „Endoscope and accessories” 123 Product Codes unter sich. Die von der FDA definierte Produktfamilie „Endoscope and accessories“ hat also 123 Mitglieder. Andere Regulation Numbers enthalten dagegen lediglich einen Product Code.
Erst auf der Ebene des Product Codes wird deutlich, welche Risikoklasse auf das Produkt zutrifft und welcher Zulassungsweg vorgesehen ist. Ebenso können auf dieser Ebene produktspezifische Normen zur Berücksichtigung angegeben werden.
Die Abbildung 1 fasst den Ansatz zur Gruppierung von Medizinprodukten der FDA grafisch zusammen.
Anschauungsbeispiel
Im Folgenden möchte ich den Klassifizierungsweg verdeutlichen an- hand eines Beispielproduktes: ein Schwamm, welcher während offen-chirurgischer Eingriffe zur Hämostase in den Körper eingebracht wird und zur kurzzeitigen Anwendung vorgesehen ist.
Der Anwendungskontext ist recht unspezifisch im Bereich chirurgischer Eingriffe zu sehen. Hier bietet sich die Medical Specialty „General and Plastic Surgery Devices” an (21 CFR 878).
In dieser Medical Speciality sind 102 Regulation Numbers enthalten. Bei der Suche nach „Sponge“ ergeben sich auf dieser Ebene zwei Treffer:
- 21 CFR 878.4014: Nonresorbable gauze/sponge for external use
► Trifft nicht zu, da eine externe Anwendung angezeigt ist - 21 CFR 878.4452: Nonabsorbable expandable hemostatic sponge for temporary internal use
► Hier sind wir auf der richtigen Spur
Unter der 878.4452 findet sich die Beschreibung dieser Regulation Number. Neben einer konkreteren Beschreibung des Produkts finden sich eine ganze Reihe von Special Controls. Zum Beispiel. müssen durch „non-clinical performance testing“ (i. d. R. sind damit Bench-Tests gemeint) unter anderem Punkte wie das Ausmaß der Ausdehnung, Absorptionseigenschaften oder mechanische Eigenschaften adressiert werden. Ebenso müssen „in-vivo performance data“ Tierversuche durchgeführt oder klinische Daten zu Punkten wie „Deployment“, „Control of Bleeding“ etc. erhoben werden.
Sucht man nun mit der Regulation Number 878.4452 in der Produktklassifizierungsdatenbank, so findet sich ein Treffer:
Product Code: PGZ
Device: Non-Absorbable, Expandable, Hemostatic Sponge for Temporary Internal Use
Device Class: 2
Submission Type: 510(k)
Ohne an dieser Stelle weiter darauf einzugehen: Die 510(k) Einreichung basiert auf der Argumentation der Vergleichbarkeit des eigenen Produkts zu einem bereits auf dem US-Markt befindlichen Produkt. Damit ist klar, dass für solche Produkte ein sog. „Predicate Device“ herangezogen werden muss.
Die Vergleichbarkeitsargumentation bietet einigen Spielraum, es sollte jedoch ein Predicate Device aus dem gleichen Product Code herangezogen werden.
Mit der Information zum Product Code können die in Frage kommenden Produkte recht schnell identifiziert werden. Die 510(k) Database kann bspw. unter Verwendung des Product Codes als Suchbegriff durchsucht werden.
Im Falle des nicht-absorbierbaren Hämostaseschwamms (Product Code „PGZ“) stehen 5 konkrete Produkte zur Auswahl. Diese sollten genau bewertet werden, um eine angemessene Auswahl eines Predicate Devices treffen zu können.
Tipps
Die oben dargestellte Herangehensweise ist eng an die Gruppierungshierarchien der FDA angelehnt. Teilweise kann die Entscheidung bereits auf Ebene der Medical Specialty uneindeutig sein. Es bietet sich daher an, die Suche nach dem richtigen Product Code aus mehreren Winkeln anzugehen. Ein ähnliches Produkt eines anderen Herstellers, welches bereits auf den US-amerikanischen Markt zugelassen ist, kann einen guten Anhaltspunkt bieten.
Um die Product Codes zu bereits vermarkteten Produkten zu finden, können Produkt- und/oder Hersteller in die „Establishment Registration & Device Listing“ Datenbank eingegeben werden.
Ausblick und Zusammenfassung
Im Gegensatz zu den europäischen Regularien arbeitet die FDA mit keinem konkreten Regelkatalog zur Klassifizierung von Medizinprodukten. Stattdessen findet eine umfangreiche Gruppie- rung von Medizinprodukten auf drei Ebenen statt:
- Medical Specialty: Grober klinisch-medizinsicher Kontext
- Regulation Number: Darstellung einer Produktgruppe innerhalb des medizinischen Kontexts
- Product Code: Spezifischer Produkttyp innerhalb einer Produkt- gruppe
Auf Ebene der Regulation Number können Special Controls beschrieben werden – diese stellen konkrete Anforderungen dar, für die Produkte innerhalb der Regulation Number adressiert werden müssen.
Auf Ebene des Product Codes wird unter anderem auch der Einreichungsweg definiert.
Ein gutes Verständnis der FDA-Gruppierung von Medizinprodukten und eine systematische Recherche potentiell passender Product Codes sind essentiell, um eine Einreichung bei der FDA richtig anzugehen.
Die FDA-Datenbanken sind untereinander verlinkt – klickt man auf „new search“, so erscheint rechts ein Auswahlfeld zu anderen FDA-Datenbanken. So können die Informationen aus einer Datenbank schnell in die Suchmaske einer anderen Datenbank eingegeben werden.
Generell empfiehlt es sich, den Umgang mit den FDA-Datenbanken zu üben. Neben dem Verständnis für die Gruppierung von Medizinprodukten und der hier beschriebenen Recherche der Regulation Number, Product Codes strebt die FDA besonders zur Auswahl des Predicate Device eine weitere Vereinheitlichung an. Derzeit befindet sich ein FDA-Guidance Dokument in Erstellung, welches „Best Practices“ zu Predicate Device Auswahl beschreibt. Zwar ist die bisher vorliegende Entwurfsversion noch nicht finalisiert, es ist allerdings erkennbar, dass die FDA bei 510(k)-Einreichungen besser nachvollziehen möchte, auf Basis welcher Kriterien ein Predicate Device zu Beginn der 510(k)-Route ausgewählt wurde. Eine entsprechende Dokumentation und Begründung der Predicate Device Suche und Auswahl ist daher zu empfehlen.
Probieren Sie es also aus und recherchieren Sie Regulation Numbers und Product Codes, die auf Ihre Produkte zutreffen könnten! Dies ist der erste Schritt zu einer erfolgreichen Zulassung ihrer Medizinprodukte auf dem US-amerikanischen Markt. Bei diesem – und allen weiteren Schritten – stehen wir Ihnen natürlich gerne mit unserem Know-How unterstützend zur Seite.
Kontakt
Amko Groeneveld, B.Sc.
amko.groeneveld@novineon.com
+49 7071 / 98979 - 147
Download

Amko Groeneveld, B.Sc.
Senior Consultant
amko.groeneveld@novineon.com
+49 7071 / 98979 - 147