
Milestones on the road to clinical evaluation

A clinical evaluation is a systematic and planned process for the continuous generation, analysis, and evaluation of clinical data on a product. It is required for all medical devices, regardless of their risk classification. The benefit-risk ratio is assessed on the basis of sufficient clinical data. According to the MDR, the clinical evaluation should be appropriate to the product, taking into account its characteristics and intended purpose. But what does 'appropriate' mean, and when is the data 'sufficient'? This article describes the six milestones for a successful clinical evaluation.
Grouping of products for clinical evaluation
The first step is to consider which products should be included in the clinical evaluation. Sometimes it is a single product, in which case the situation is clear. However, it often makes sense to evaluate several products together in a clinical evaluation.
For large product groups based on the same technology and with a shared purpose, joint assessment is a good option. If the reporting processes - i.e., risk management and post-market surveillance processes - are also coordinated, many redundancies can be eliminated (see below).
In other cases, products are used together in everyday medical practice, forming a combination of effects . In such systems, the desired medical effect cannot be achieved by the individual products alone. Consequently, the performance and safety of these products can only be evaluated and demonstrated in combination. the desired medical effect cannot be achieved by the individual products alone. In such cases, grouping may be appropriate even if the products represent different technologies and belong to different risk classes. In this case, the procedure would be based on the product with the highest risk class. This may result in documents such as the PSUR being created “unnecessarily” or “too frequently” for some products. However, this makes the overview and argumentation much easier.
Positioning – Which product category should I compete in?
Right at the outset, it is important to clarify which data and data sources are available. However, the exact requirements for clinical evaluation depend on the product's risk class.
The products can be divided into two broad categories: high-risk products, i.e., Class III and implants, and low-risk products, i.e., non-implantable products in Classes I to IIb.
Clinical trials are required for high-risk products under Article 61(4). For low-risk products, however, no explicit statement is made. Nevertheless, the clinical evaluation must be based on 'sufficient clinical data'. Whether this is the case is assessed on a case-by-case basis.
However, MDR 2 (48) provides a precise definition of 'clinical data'. According to the MDR, only data collected with the actual product, or an equivalent product is considered “clinical data”. Therefore, data on the generic product group is not considered “clinical data”. Nevertheless, according to MDCG document 2020-6, such data may also be used in cases of well-established technology. Therefore, fundamental considerations regarding equivalence assessments and critical evaluation of the available evidence must be made.
Choice of clinical evaluation strategy
There are several routes available for clinical evaluation. Before preparing the clinical evaluation, manufacturers should consider the following questions:
- What empirical values/data are available?
- Can the risk-benefit ratio be defined; are there any uncontrolled residual risks?
- Which questions regarding the performance, safety, and clinical benefits of the product remain unanswered?
- Can the data gap be closed using preclinical methods, or is clinical data required?
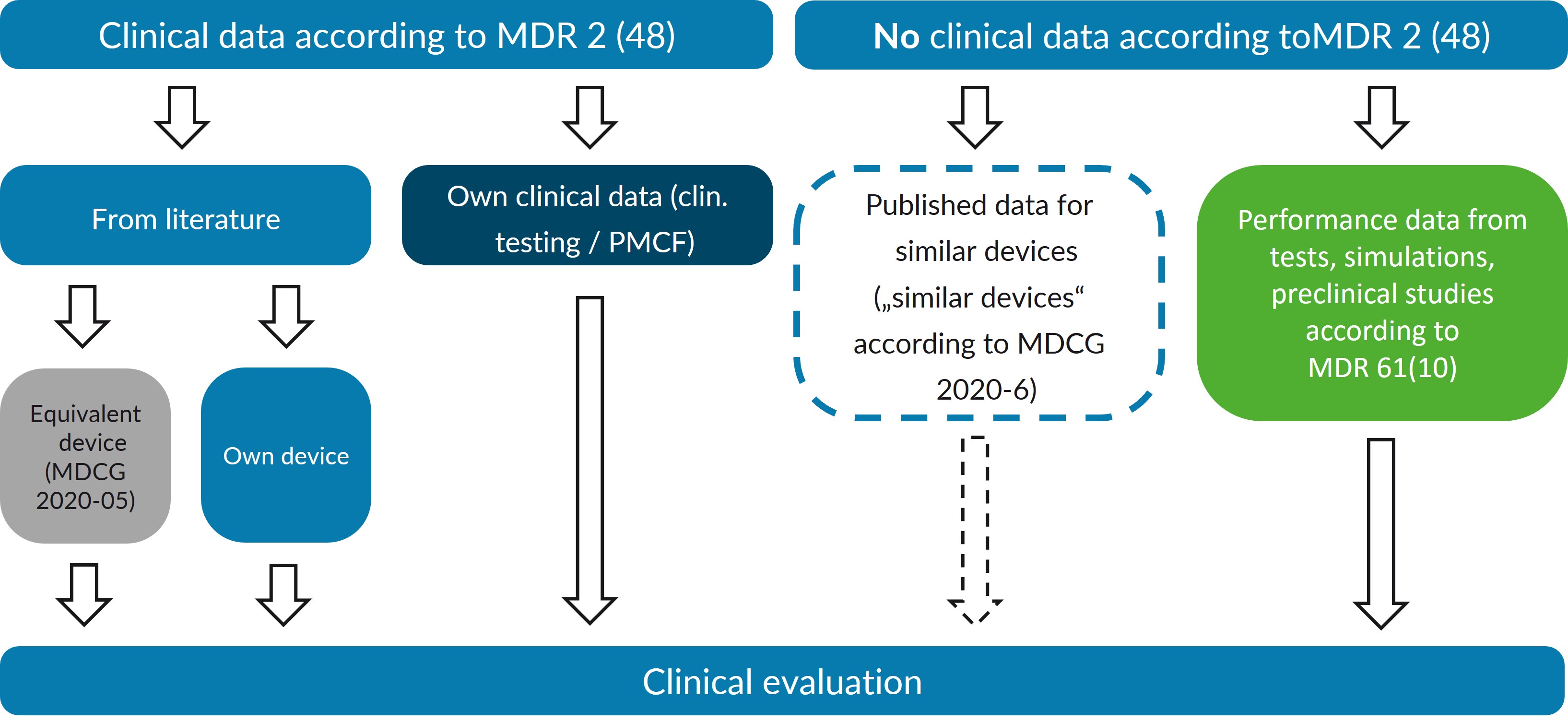
Direct evidence: Own data
The MDR establishes a certain hierarchy in the definition of clinical data. Clinical trials of the product in question are given priority. Along with clinically relevant information from post-market clinical follow-up (PMCF), this data constitutes the 'direct evidence' for the product. This data can be published in medical journals or be proprietary clinical data.
Indirect evidence: Equivalence
Product with external equivalence according to MDR 61(5)
The equivalence route, which used to be the main form of clinical evaluation, has many disadvantages under the MDR. For one thing, this route is “regulatorily unstable.” This is because if the intended purpose of the equivalent product changes—for example, if indications are removed or contraindications are added—lines of argumentation in the clinical evaluation break down.
On the other hand, the MDR aims to encourage manufacturers to move away from the equivalence route wherever possible and to demonstrate conformity with the essential safety and performance requirements (ESPR) using direct clinical evidence. As a result, the equivalence criteria have become more stringent under the MDR, irrespective of the risk class (MDR, Annex XIV, Part A(3) and MDCG-2020-5).
In addition, sufficient access to the information necessary for equivalence assessment is required. The MDR requires 'unrestricted access to technical documentation' for 'high-risk products', which is only available in the rarest of cases. But even for “low-risk products,” publicly available information may not be sufficient.
Product with equivalence to a device already marketed by the same manufacturer, according to MDR 61(4)
For high-risk products in particular, it may be advisable to opt for the route of internal equivalence in the clinical evaluation. This involves postulating equivalence to a predecessor product or a similar product from the company's own portfolio. If the equivalence criteria can be met, this is a good way to bring new products into the MDR.
Performance data 61(10)
The MDR acknowledges the possibility that “clinical data” may not be suitable for demonstrating a product's conformity with the GSPR. In this case, the clinical evaluation is instead based on performance data obtained from testing, simulations, or preclinical trials. This approach is particularly suitable for simple products that do not fulfill an independent medical purpose (such as equipment) or whose function can be better tested in animal models or bench tests than in clinical studies. It is important that no direct clinical benefit - e.g., improved safety or performance - may be postulated for these products.
The performance data route is only open to “low-risk products.” This is because Article 61(10) clarifies that Article 61(4) takes precedence, requiring clinical investigations for Class III products and implants.
Simple implants: Well-established technology 61(6b)
Another exception where “clinical data” can be waived in accordance with the MDR is for products in the “well-established technology” group. The MDR specifies simple implants such as screws, plates, and suture material (MDR 61 (6b)). The MDCG 2020-6 document adds that other products may also be considered “well-established technology” if they meet certain criteria (see MDCG 2020-6, Section 1.2). Data on similar devices are also relevant for the clinical evaluation of these products, even if they do not meet the definition of “clinical data” according to MDR 2 (48).
Legacy device 61(6a)
Legacy devices that have already obtained CE marking under the MDD constitute another exception. According to MCDG 2020-6, post-market clinical data, together with the clinical data generated for conformity assessment under the MDD/AIMDD, should form the basis for clinical evaluations under the MDR.
Clinical evaluation plan
The clinical evaluation plan classifies the product from regulatory and clinical perspectives, defining the evaluation strategy. One of the biggest challenges in creating this plan is defining the safety and performance parameters to be used to evaluate the product and its risk-benefit ratio. Ideally, these parameters can be derived from the state-of-the-art literature, are quantifiable, and serve as endpoints in published scientific studies. In this case, the performance and safety of the product in question can be easily compared with the state of the art.
Unfortunately, the ideal scenario rarely occurs. Often, the parameters that can be used to meaningfully measure the safety and efficacy of a product are not the focus of clinical research. In such cases, it is impossible to compare data on the product itself or on comparable products and alternative treatments. For such products, it is therefore particularly important to carefully define the evaluation parameters and methods for such products so that the questions posed can be answered.
Coordinate processes
Clinical evaluation is a key process for identifying and analyzing clinical data, demonstrating that a medical device conforms to the Essential Safety and Performance Requirements (ESPRs). During this systematic assessment, gaps in clinical data may be identified that reveal unanswered questions about the product's performance or safety profile.
In addition, clinical evaluation can identify potential deficiencies in the risk management documentation. Any gaps or uncertainties identified must be systematically addressed through well-designed post-market surveillance (PMS) and post-market clinical follow-up (PMCF) plans. As PMS and PMCF activities generate new clinical insights and evidence from practice, these findings must be incorporated into subsequent clinical assessments, creating a continuous feedback loop.
This new information may necessitate an updated benefit-risk assessment and prompt corresponding changes to the risk management documentation. This interconnected, cyclical approach ensures that clinical evaluation, post-market surveillance (PMS), post-market clinical follow-up (PMCF) and risk management work together to maintain the safety and performance of the medical device throughout its lifecycle, meeting both regulatory requirements and the product's intended purpose.
To ensure this system functions optimally, careful timing of the various reports and plans is essential so that the insights gained can be incorporated into the subsequent processes in a timely manner and regulatory deadlines can be met.
Establishing a PMCF strategy
Post-market clinical follow-up (PMCF) is an essential activity under the MDR. PMCF provides an important source of proprietary clinical data for future conformity assessment procedures. For products that have obtained CE marking using the equivalence route, PMCF should aim to become independent of this data set. This is because if problems arise with the equivalent product or it is withdrawn from the market, one can quickly find oneself at a disadvantage when it comes to evaluating one's own product.
All set?
Clinical evaluation brings together technical data, published literature, risk management, and post-market surveillance. It is a dynamic process that ensures the safety and performance of medical devices throughout their entire life cycle. The six milestones presented—from product grouping and strategy selection to PMCF planning—form a structured framework. The key is to integrate clinical evaluation, risk management, and post-market surveillance into a coherent system. Manufacturers who follow this approach not only meet regulatory requirements, but also gain valuable insights for continuous product improvement and ultimately strengthen the benefit-risk ratio of their products for the benefit of patients.
