
How to avoid common problems in the IVDR conformity assessment procedure
Expert tips

Are you, as an IVD manufacturer, currently in the midst of the demanding process of CE certification or preparing for it? Have you already encountered one or more hurdles while implementing the requirements of the IVDR? A recent survey by the EU Commission among Notified Bodies shows: submissions are largely assessed as incomplete. We do not assume that manufacturers intentionally submit with glaring gaps. However, it is clear that manufacturers and notified bodies have different interpretations of what constitutes "completeness.". Below, we show you ways to ensure the completeness of your submitted files and facilitate the notified body's review.
Despite extended transition periods, address the switch to IVDR early
Regulation (EU) 2017/746, hereinafter referred to as the IVDR, has been in force since May 26, 2017. Regulations 2022/112, 2023/607, and 2024/1860 extended the transition periods (see also our technical information "Further extension of the transition periods for IVDs") . Our urgent recommendation to all IVD manufacturers: make the most of the time gained! Not only IVD manufacturers but also notified bodies report major challenges posed by the IVDR requirements.
Notified bodies report incomplete files as the main problem
The entry into force of the IVDR has not only brought about significant changes for manufacturers (see Table 1 for further details); notified bodies are also facing new challenges. For example, the introduction of the revised classification system is leading to more product certifications involving notified bodies.
Tabel 1: Overview of the changed requirements due to the IVDR
- New / Increased Requirements According to IVDR
- Risk-based classification system A-D. In addition, further types of IVDs are mentioned (point-of-care tests, products for self-testing, and companion diagnostics). Furthermore, the manufacture and use of in-house IVDs (also called lab-developed tests or LDTs) are now subject to the requirements of the IVDR.
- The conformity assessment procedure now requires the involvement of a Notified Body in many cases. Only manufacturers of non-sterile Class A products are still allowed to self-declare conformity.
- Quality management according to IVDR Article 10 and ISO 13485.
- Product-specific technical documentation according to IVDR Annex II.
- Post-market surveillance according to IVDR Annex III.
- The IVDR places particular emphasis on performance evaluation and requires continuous monitoring of performance and risks throughout the entire product lifecycle. Post-market performance follow-up (PMPF) is also required.
- In addition to harmonized standards, there are “Common Specifications” for high-risk products. These define specific criteria for safety and performance requirements.
- Detailed requirements for information provided by the manufacturer, including labeling and instructions for use according to IVDR Annex I Chapter III.
- The IVDR addresses the topic of software much more extensively than the IVDD. Software verification and validation are required, as well as ensuring IT security of the products. Software must be labeled according to the UDI system. There are also special classification rules for software.
- For clear product identification, IVDs must be labeled according to the UDI system and the corresponding information must be stored in the EUDAMED database.
- Designation of a “Person responsible for regulatory compliance (PRRC).” Manufacturers must appoint at least one employee in their company to ensure compliance with the requirements.
- Additionally, the requirements for documentation and conduct of (clinical) performance studies have multiplied.
To monitor and analyze the availability of medical devices and in vitro diagnostics on the EU market, in connection with the implementation of the regulations on medical devices and in vitro diagnostics from the perspective of key stakeholders, the EU Commission has commissioned a data collection study. This study is planned for 36 months, starting in December 2022. As part of the project, a dashboard has been set up that compiles the latest data. An overview of the study and the study-related dashboard can be found here: Study supporting the monitoring of availability of medical devices on the EU market - European Commission (europa.eu).
In the area of IVDR, statistics on submissions and certifications, their quality and duration, IVDR codes, etc. are shown.
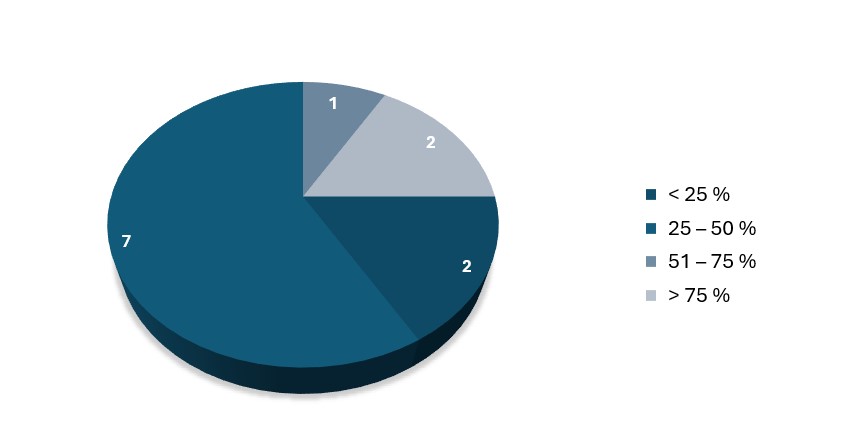
In the area of quality and processing time on the dashboard, the current status (as of February 2024) is as follows: 9 out of 12 Notified Bodies report that more than 50% of submissions are incomplete. Only 2 out of 12 Notified Bodies report that more than 75% of submissions are complete (see Figure 1). In addition to the incompleteness of the submitted files, other reasons for the rejection of submissions are cited, such as incorrect classification of the submitted products, incorrect conformity assessment procedure, and other unspecified reasons. From our perspective, the information is often available, but not in the form that the Notified Body expects or requires.
Regarding the processing time, 9 out of 12 Notified Bodies report that the conformity assessment procedure takes 13–18 months, one out of 12 Notified Bodies indicates a period of 6–12 months, and for two out of 12 Notified Bodies, it can even take up to 24 months before manufacturers can expect issued QMS or product certificates. These long review times “freeze the file.” Product-specific information, as well as formal aspects such as newly published MDCG documents, cannot be incorporated into a file that has already been submitted.
Conclusion
Notified Bodies report two main problems with conformity assessment procedures:
- The completeness of the submitted files
- The time required to obtain the certificate
How can manufacturers more easily compile all the information and documents required for submission and efficiently guide the Notified Body through the document jungle, without additional documentation effort?
Use checklists as key documents for dossier submission!
The MDCG (Medical Device Coordination Group) is intended to provide support. It addresses the most important questions regarding medical devices, including in vitro diagnostics, and publishes the relating MDCG documents. These provide a general understanding of how the MDR and IVDR should be applied in practice to achieve effective implementation of the regulations. In the IVD sector, MDCG documents already exist on the following topics:
- Extension of transition periods (MDCG 2024-4; MDCG 2022-15; MDCG 2022-6; MDCG 2021-4)
- Classification of IVDs (MDCG 2024-11; MDCG 2020-16)
- Performance studies (MDCG 2024-4, MDCG 2022-20; MDCG 2022-19, MDCG 2022-10)
- Expert panels for class D products (MDCG 2021-22, MDCG 2022-3)
- Clinical evidence (MDCG 2022-9; MDCG 2022-2)
- Legacy devices (MDCG 2022-8)
However, these documents are not legally binding and so far, only cover a few of the new requirements introduced by the IVDR. As a result, many specific questions from manufacturers remain unanswered. Although manufacturers and Notified Bodies interpret and implement the IVDR to the best of their knowledge and belief, their interpretations sometimes differ significantly.
Some challenges cannot be solved in the short term, such as the publication of clarifying MDCG documents. Other solutions, such as the use of checklists, are already available. For self-checks, manufacturers can go through Annex II step by step and verify whether all points are addressed in the submitted file. Notified Bodies have also recognized this and, as a helpful resource, provide checklists and best practices for completeness checks publicly on their websites or upon request. This is a structured preparation of Annex II of the IVDR.
Our tip
Use the checklist provided by your Notified Body or Annex II. Attach the TD checklist to your submitted file so that your Notified Body can immediately see which documents are included.
This way, you know that your file is complete, or you can identify where there are still gaps and which points need further revision. However, this does not necessarily mean that your file is complete in terms of content, or that conformity with the General Safety and Performance Requirements (GSPRs), as required in Section 4 of Annex II of the IVDR, can be demonstrated. In addition, justifications for the solutions chosen to fulfill the GSPRs, as well as their validation and verification, must be provided. Annex I sets out the General Safety and Performance Requirements that apply to the product, taking into account its intended purpose.
Use Annex I as a template and work through it step by step. By using this so-called GSPR checklist, you can efficiently guide yourself and your Notified Body through the necessary decision points. This clearly shows which GSPRs apply to your product and provides a justification for why certain requirements may not be applicable.
Conclusion
You know your product and therefore the technical documentation best! Attach the completed TD checklist to your submitted file and demonstrate that your file formally addresses all points according to Annex II of the IVDR. This way, your submission will be among the 75% that are rated as complete in the future!
With a carefully completed GSPR checklist, you can efficiently guide the Notified Body through your submitted file and save valuable time. Use the language of the IVDR so that your Notified Body can more easily find the relevant IVDR terms. These IVDR terms should be used consistently to prevent inconsistencies.
We are, of course, happy to support you in this process. Often, the information is already available - it just lacks the link to IVDR terminology. We are happy to help you make these connections!
